library(tidyverse)
library(wastewateR)Processing Vignettes
Vignette #1 - A Brief Overview
This vignette shows an example of how to string together the processing stand-alone and wrapper functions to process a single output file from a wastewater laboratory run. In instances where it makes sense to dive deeper into how the functions work, that detail will be included. However, other vignettes may cover specific scenarios in more detail.
First - Know Your Data
This processing system assumes that the raw wastewater data pulled off the laboratory machines is un-merged, as in, the values for all replicates for each sample is included in the output.
Depending on the output format from the machine and software in a particular lab, the output columns may not align with the expectations of the functions included here. There is one function software_renaming() that can assist with this. Regardless, the wrapper processing functions expect an input with the following columns:
- “Well”, “Sample”, “Target”, “Concentration”, “CopiesPer20uLWell”, “AcceptedDroplets”, “Positives”, and “Negatives”
The data file off the machine in the lab may have columns including:
- “Well”, “Sample.description.1”, “Target”, “Conc.copies.µL.”, “Copies.20µLWell”, “Accepted.Droplets”, “Positives”, and “Negatives”
# read in raw data file off the machine
raw_data_file <- read.csv("{data_file_name.csv}")
# it is recommended to store the raw data file for record keeping
# take a quick look at the file
head(raw_data_file)
# if there is a column called "Sample.description.1" in the raw data file
# want to use the software_renaming() function
if (any("Sample.description.1" %in% colnames(raw_data_file))){
# this new dataframe, format for use, will have only the 8 columns the
# processing functions are expecting
format_for_use <- software_renaming(raw_data_file, style = "qx_manager")
}| Raw Data File | Transformed To | Definition |
|---|---|---|
| Well | Well | The well the sample was in, such as A01 |
| Sample.description.1 | Sample | Identifying sample name, all replicates should have the same sample name |
| Target | Target | The target the well is being tested for |
| Conc.copies.µL. | Concentration | Concentration value in copies per uL |
| Copies.20µLWell | CopiesPer20uLWell | Copies per 20uL value in well |
| Accepted.Droplets | AcceptedDroplets | Count of accepted droplets, sum of Positives and Negatives |
| Positives | Positives | Count of positive droplets detected |
| Negatives | Negatives | Count of negative droplets detected |
post_processing <- qaqc_processing_sc2_n1(file_in = format_for_use,
lab_site_ids = c("{id}", "{id}"))The processing minor and wrapper functions are quality control, quality assurance (QAQC) type functions. There is messaging that will output as the function runs, to indicate information about the data. The end output is a list, where the first element is a dataframe of processed data, and the second element is a dataframe of what may be considered “stop error” notifications.
If the messaging and stop error notifications meet the users’ specifications for continuation, they may still want to process the data further into a metric such as “gene copies per 100mL”.
concentration_data <- read.csv("{data_file_name.csv}")
# this data frame is expected to have "Sample", "initial_volume_analyzed_mL", "final_concentrate_volume_mL" as its three columns.
measurement_values <- calculate_gc_per_100ml(lab_df_in = post_processing[1][[1]],
all_weigh_info = concentration_data)
# "detection_limit_CP_100mL" and "CP_100_mL_of_sample" are calculated, following MDHHS
# SEWER methodologyVignette #2 - Deeper Dive into Wrappers & Minor Functions
This vignette dives into an example of how the wrappers and minor functions of this package interact.
The wrapper functions are all intended as processing functions for a specific pathogen or set of pathogens. The idea is that the majority of the standard inputs and rules are set, and the user just needs to provide a small amount of information that is specific to their use-case (i.e. their data file, and their sample site identifiers). Most wrappers include additional flexibility, including the ability to change control naming conventions. If a user wanted additional flexibility beyond that, then they would need to string together the minor functions for their own use.
What Happens Before Using wastewateR
Laboratory methods (RNA/DNA extraction, ddPCR)
Thresholding
Final results data file export (likely from Quantasoft BioRad Software)
Figure 1. An Explanation of the SARS-CoV-2 N1 ‘Slim’ Wrapper Function 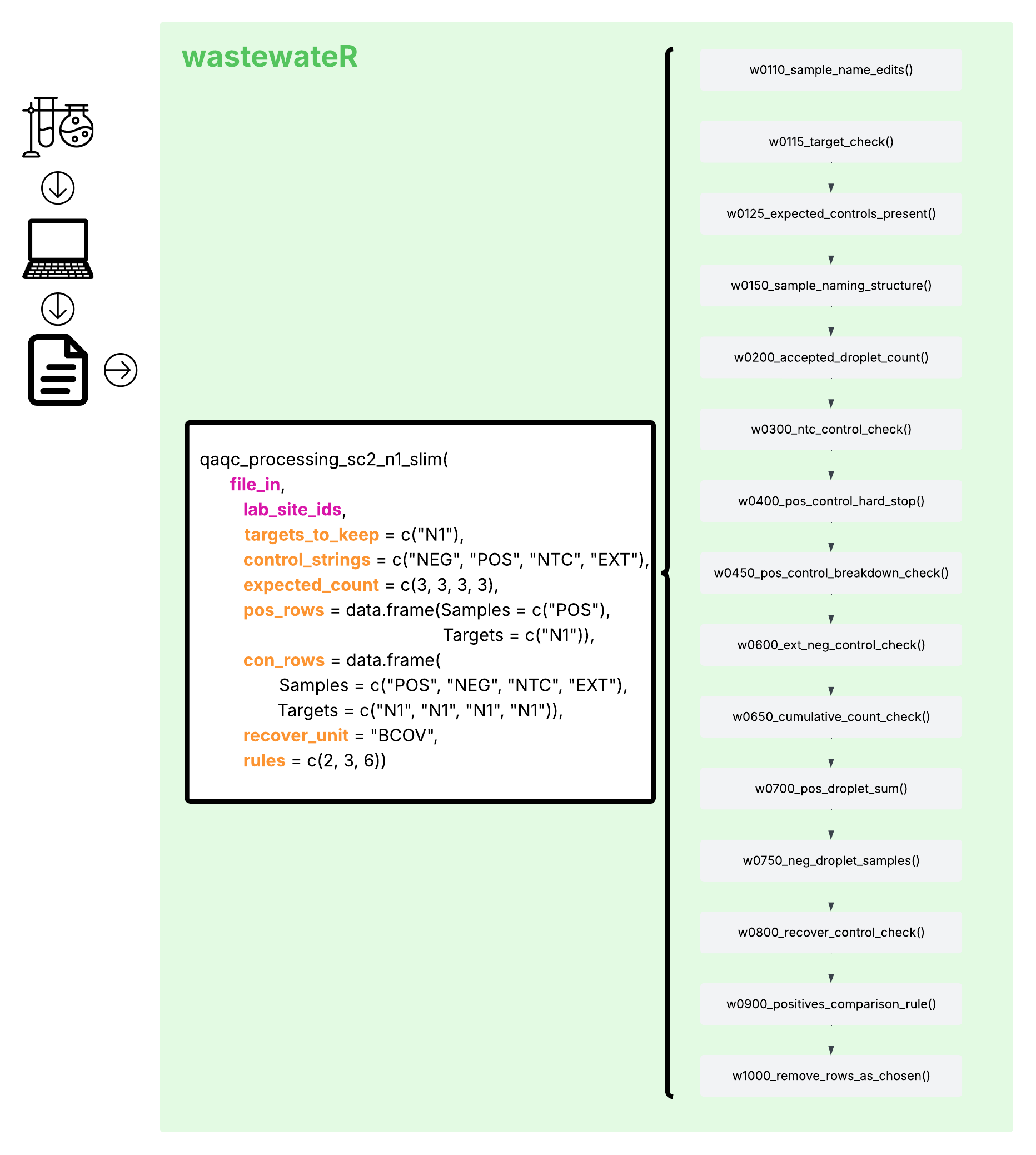
Using wastewateR for Processing / QAQC
For processing / post-laboratory methods and pre-analysis quality assurance & quality control (QAQC), the wastewateR package is meant to handle a .csv file export of laboratory data results. The main source of that file is expected to be the Quantasoft BioRad Software, however, the columns are fairly consistent with other measurement expectations, so other sources should be able to be processed.
There are stand-alone functions that are part of wastewateR, however, those are described elsewhere. For this vignette, we are focusing on the interaction between the minor functions and the wrapper functions, within the context of processing and QAQC. The goal of wastewateR was to be modular and flexible, while still handling the standardization and automation ideals for higher quality data processing within the context of wastewater epidemiology. There are other use cases for this package, but this was the impetus for its creation. There was an understanding that different laboratories and different testing methods would require different standards and inputs. The package was therefore, built modularly (i.e. the minor functions exist) but with an eye for ease of use, these minor functions were put into a set of “standard” functions, which here we call wrappers. These attempt to provide a scaffolding to cover the most likely situation for different pathogens. Please also note, that the initial creation of this package was for a specific wastewater epidemiology program at the state of Michigan’s Department of Health and Human Services, so there are some features that reflect their standards and would perhaps need alteration for others to use.
In Figure 1, the fourteen minor functions that are used within the wrapper function are displayed in grey, in the order that they are used. The wrapper function name and the inputs that it allows are in the white square to the left of those listed functions. In that white square, the inputs that are REQUIRED by the function are written in bold hot pink. All other inputs are written in bold orange and are not required for use. If they are not provided, the default inputs are used. However, their presence allows for some additional flexibility within the use of the wrapper function, hopefully decreasing some need for stringing the minor functions together oneself.
The wrapper function:
qaqc_processing_sc2_n1_slim <- function(file_in,
lab_site_ids,
targets_to_keep = c("N1"),
control_strings = c("NEG", "POS", "NTC", "EXT"),
expected_count = c(3, 3, 3, 3),
pos_rows = data.frame(Samples = c("POS"),
Targets = c("N1")),
con_rows = data.frame(Samples = c("POS", "NEG", "NTC", "EXT"),
Targets = c("N1", "N1", "N1", "N1")),
recover_unit = "BCOV",
rules = c(2, 3, 6)){
file_in <- w0110_sample_name_edits(file_in)
file_in <- w0115_target_check(file_in, targets_to_keep)
error_line <- c("w0115")
error_val <- c(file_in[2][[1]])
file_in <- w0125_expected_controls_present(file_in[1][[1]],
control_strings,
expected_count)
error_line <- c("w0125")
error_val <- c(file_in[2][[1]])
file_in <- w0150_sample_naming_structure(file_in[1][[1]],
lab_site_ids,
control_strings)
error_line <- c(error_line, "w0150")
error_val <- c(error_val, file_in[2][[1]])
file_in <- w0200_accepted_droplet_count(file_in[1][[1]])
file_in <- w0300_ntc_control_check(file_in)
error_line <- c(error_line, "w0300")
error_val <- c(error_val, file_in[2][[1]])
error_line <- c(error_line, "w0400")
error_val <- c(error_val, w0400_pos_control_hard_stop(file_in[1][[1]], pos_rows))
file_in <- w0450_pos_control_breakdown_check(file_in[1][[1]], pos_rows, 35)
file_in <- w0600_ext_neg_control_check(file_in, 3, 3, 3, 1)
error_line <- c(error_line, "w0600")
error_val <- c(error_val, file_in[2][[1]])
error_line <- c(error_line, "w0650")
error_val <- c(error_val, w0650_cumulative_count_check(file_in[1][[1]], 3, "no"))
file_in <- w0700_pos_droplet_sum(file_in[1][[1]], controls_to_drop = con_rows)
file_in <- w0750_neg_droplet_samples(file_in, controls_to_drop = con_rows)
error_line <- c(error_line, "w0750")
error_val <- c(error_val, file_in[2][[1]])
file_in <- w0800_recover_control_check(file_in[1][[1]], recover_unit, control_strings, 0.3)
file_in <- w0900_positives_comparison_rule(file_in, pos_rows[, 1], pos_rows[, 2], control_strings, 3)
file_in <- w1000_remove_rows_as_chosen(file_in, rules)
error_df <- data.frame(error_line, error_val)
message("End wrapper for SARS-CoV-2 N1 (slim).")
return(list(file_in, error_df))
}![]()